
The chimera mystery#
Direct RNA sequencing (dRNA-seq) on Oxford Nanopore looks, on paper, like a transcriptomics dream. You sequence native RNA molecules end to end, you keep the modifications, and you skip every reverse-transcription and PCR step that has been quietly polluting short-read data for years. For a while, that was the story we were telling ourselves.
Then the multi-mapped reads showed up.
When we aligned VCaP dRNA-seq data with minimap2, a non-trivial fraction of reads came back as chimeric alignments: two distant pieces of the genome stitched together by what looked, at first glance, like a fusion transcript.
A 2020 paper had hinted that some of these were artifacts, not biology, but nobody had systematically pinned down where they came from, how often they occurred, or how to fix them.
We could not tell whether we were looking at exciting prostate-cancer fusions or expensive garbage.
That ambiguity is what eventually became DeepChopper.
Why the existing tools could not help#
The first thing I tried, and you should always try this first, was to reach for an existing tool. There are good ones for cDNA: Pychopper, Porechop, Porechop_ABI. ONT’s own basecaller, Dorado, even has built-in adapter trimming.
On dRNA-seq, all of them collapsed to near-zero F1 for the case we cared about: internal adapters. The reason was subtle. Pychopper looks for cDNA-specific primers that simply don’t exist in dRNA-seq. Porechop and Porechop_ABI rely on exact k-mer matches or stable k-mer signatures. Dorado was built around 3′-end trimming, not arbitrary internal junctions. And, critically, the dRNA basecaller is trained on RNA. When an ONT adapter (DNA) sits inside a read, the RNA model basecalls it as a low-quality, fuzzy mess that bears only a statistical resemblance to the canonical adapter sequence.
In other words: the artifact has a fingerprint, but the fingerprint is corrupted, and corrupted fingerprints break every exact-matching tool by construction.
The insight#
The breakthrough was reframing the question. Instead of asking “is this an alignment artifact?”, we asked “is there an adapter inside this read?” Adapter-bridged chimeras carry an internal adapter; biological chimeras don’t. If we could read corrupted adapters out of noisy basecalls, we could distinguish artifacts from real RNA rearrangements without ever touching the alignment.
That meant we needed something that:
- Scans full transcripts (not 512 bp windows),
- Tolerates a lot of sequencing noise,
- Predicts at single-nucleotide resolution, and
- Doesn’t require alignment.
This is exactly the regime where genomic language models (GLMs) shine.
Why a genomic language model#
I’d been watching GLMs evolve for a while, and the context-window numbers tell most of the story:
| Model | Max input | Parameters |
|---|---|---|
| DNABERT | ~512 bp | ~110 M |
| DNABERT-2 | ~10 kb | ~117 M |
| Nucleotide Transformer | ~6 kb | up to 2.5 B |
| Evo | very long | billions |
| HyenaDNA | 32 kb | ~6.6 M (base) |
We needed at least mRNA-scale context. Human protein-coding transcripts have a median of about 2.7 kb, and the 99th percentile sits near 14 kb. HyenaDNA’s 32 kb window covered >99.97% of human transcripts in current annotations, and we confirmed the same in our own dRNA-seq datasets.
HyenaDNA was also small enough, under 10 M parameters, that we could fine-tune it on a single workstation and deploy it without a billion-parameter inference bill. After we added our task-specific layers, the final DeepChopper had 4.6 M trainable parameters. That ratio of capability to size mattered more than I expected.
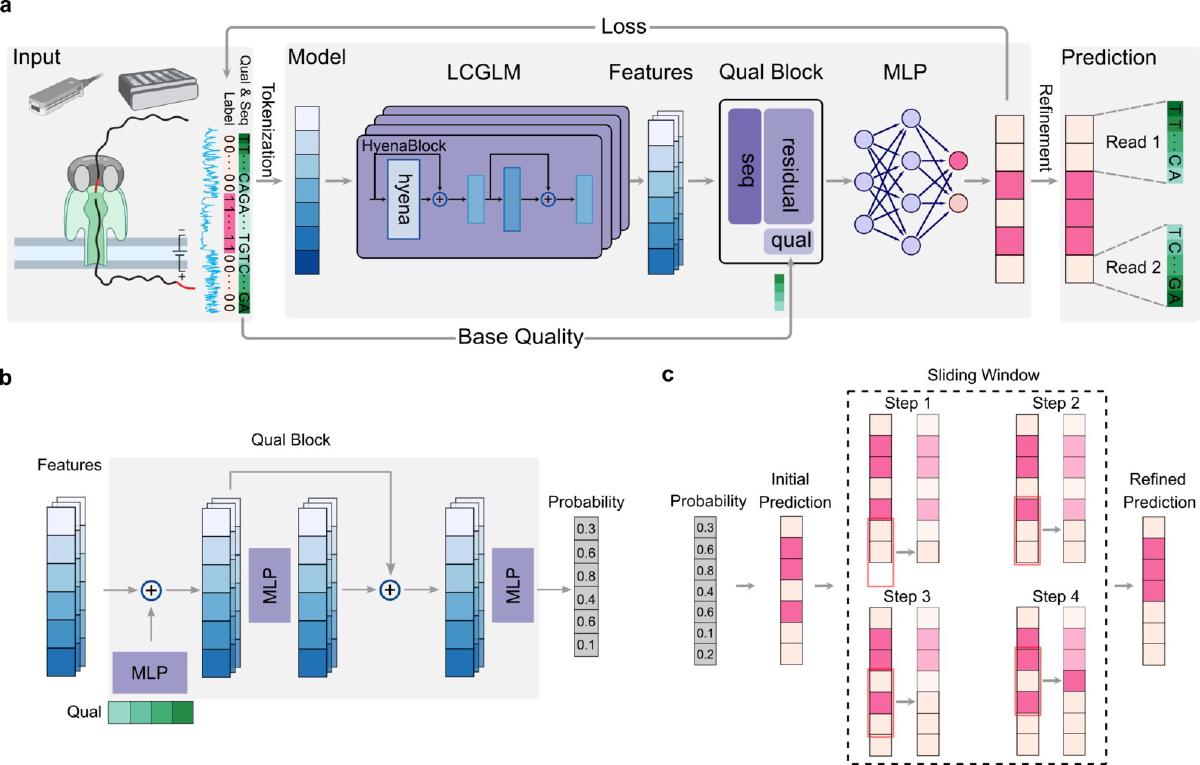
Architecture, in plain English#
DeepChopper is, mechanically, a token classifier. Each nucleotide gets a binary label: adapter or not adapter. The pipeline goes:
- Tokenize at single-nucleotide resolution (
A,C,G,T,N). - HyenaDNA backbone turns those tokens into 256-dim feature vectors with a long receptive field.
- Quality block, a small stack of MLPs with residual connections, folds in z-score-normalized per-base quality scores.
- Classification head projects each position to two logits and softmaxes.
- Sliding-window majority vote post-processes raw predictions to clean up boundaries.
The probability for nucleotide \(i\) belonging to class \(c\) is the standard softmax,
$$ P(y_i = c) = \frac{e^{z_c}}{\sum_{j=1}^{2} e^{z_j}}, $$
and we train with binary cross-entropy averaged over every base in every read of the mini-batch:
$$ \mathcal{L}{\textrm{BCE}} = -\frac{1}{N} \sum{i=1}^{N} \big[y_i \log(\hat{y}_i) + (1 - y_i) \log(1 - \hat{y}_i)\big]. $$
The quality block looks like a small detail, but it isn’t. An ablation showed F1 climbing from 0.97 → 0.99 when Q-scores were folded in. That last 0.02 buys you the difference between “good model” and “actually deployable on real data”. Basecalls inside artifacts have systematically lower Q (mean ~9.96 in our hands), and the model uses that signal to discriminate corrupted adapters from genuinely low-information regions of the read.
Engineering for scale#
A model is only useful if you can run it on a real lane of data without setting your cluster on fire. Our VCaP dataset alone was ~9 M reads; a merged 5-cell-line benchmark hit 23 M.
Three engineering decisions made that tractable:
- Rust core. FASTQ → Parquet conversion, sliding-window post-processing, and chunked I/O are all in Rust. Parquet gives us columnar batched reads instead of streaming millions of records line by line.
- GPU inference, parallelized. The HyenaDNA forward pass dominates wall time; we batch it on GPU and parallelize the surrounding stages.
- Linear scaling. End-to-end runtime on the 23 M-read benchmark was ~10.6 hours on 2× NVIDIA A100 (23 min FASTQ conversion, 8.5 h prediction, 1.7 h post-processing). Memory peaked around 56 GB on GPU and ~93 GB on CPU. Crucially, both runtime and memory stayed near-linear from 0.1 M to 23 M reads, with no obvious wall in front of larger datasets.
If you have ever had a “smart” tool quietly OOM after hour 16 of a run, you know why this matters.
Curating training data without ground truth#
One of the harder problems was honest: nobody hands you a labeled dataset of “internal adapters in dRNA-seq”. We had to build one.
The trick we converged on:
- Take primary-only alignments.
- Look at 3′-end soft-clipped regions; they are candidate adapter sequences.
- Anchor on biology. A polyA tail at the start of the soft-clip is a strong signal that what follows is the adapter, not an alignment artifact. We only kept sequences downstream of a polyA.
- Use the rest of the aligned read as labeled non-adapter.
- Synthesize chimeras by combining two non-adapter reads with one adapter, in a 1:1 ratio of internal vs. 3′-end placements, and a 9:1 positive-to-negative ratio overall.
That gave us 600,000 examples, split 8:1:1 into 480 K train / 60 K validation / 60 K test. Anchoring on polyA was the single biggest lever for label quality. Without it, the model started learning to flag anything weird as “adapter”.
What we found in the wild#
Once DeepChopper worked on synthetic data (recall, precision, F1 all > 0.99), the more interesting result came from pointing it at real datasets.
- Internal adapters in 0.33–1.62% of reads across human cell lines and chemistries. Tens to hundreds of thousands of affected reads per experiment.
- 63–85% of all chimeric reads in dRNA-seq are adapter-bridged artifacts, the dominant source of false RNA “rearrangements”.
- On VCaP RNA002, DeepChopper reduced unsupported chimeric alignments by ~95% and lifted the cDNA-supported fraction from 5.8% → 48.7%.
- The mitochondrial chromosome is a hotspot. Chimeric junctions per base are dramatically enriched on Chr M; we still don’t fully understand why, but the pattern persisted across both RNA002 and the new RNA004 chemistry.
- RNA004 didn’t make the problem disappear. The new chemistry roughly halves the artifact rate, but adapter-bridged chimeras are still systematically present. A zero-shot RNA002-trained model still cleaned them up; a small fine-tune squeezed out another 3–4%.
That last point matters because it tells you what GLMs are actually good for: generalizing across protocol shifts without recalibration. Exact-matching tools have to be re-tuned every time ONT changes a kit.
Downstream impact#
Cleaner reads only matter if they make downstream science better. Two results made me believe this was worth publishing:
- Transcript annotation. Running IsoQuant on DeepChopper-corrected VCaP reads roughly doubled the number of identified transcripts compared to uncorrected data, with the largest gains in full splice matches and alternatively spliced isoforms.
- Gene fusion detection. FusionSeeker called 89% fewer fusions on corrected data, and the calls that disappeared were precisely the ones not corroborated by short-read fusions from Arriba. False positives, gone.
The most concrete example we worked through was an apparent RPS29–COX8A fusion. The chimeric junction looked plausible by alignment alone. But when we lined up the predicted internal adapter with the raw current trace, the “fusion” sat right on a low-intensity region flanked by polyA and open-pore signals: a textbook adapter, not biology. Ribosomal-protein genes turned out to dominate the false-fusion list across every cell line we looked at.
Lessons I keep coming back to#
A few things from this project changed how I think about ML for sequencing.
Recover, don’t remove. Our first instinct was to filter chimeric reads. The much better move was to split them at the predicted adapter and recover both halves. We were leaving real biology on the floor every time we just dropped a read.
The 21 bp window was the surprise. I assumed smaller post-processing windows would always be better; they are, in raw cDNA-support percentage. But they also fragment reads into 4+ pieces too eagerly. 21 bp was the boring middle that won the sweep. Boring middles win a lot.
Small models, well-targeted, beat big models thrown at the problem. A 4.6 M-parameter model dedicated to one task outperformed everything we benchmarked, including methods that lean on much larger backbones. GLMs are not just “DNA GPT”; the architectural choices that matter are context length, tokenization granularity, and how cleanly the task maps onto token classification.
Quality scores are an under-used signal. That F1 jump from 0.97 to 0.99 came from a handful of MLP layers reading per-base Q. If you are training models on basecalls and ignoring Q, you are leaving a free 1–2 points on the table.
Try it#
DeepChopper is open source. If you sequence dRNA-seq data, RNA002 or RNA004, there is a reasonable chance some non-trivial fraction of your “fusions” are adapters in disguise. It only takes one run to find out.
- GitHub: github.com/ylab-hi/DeepChopper
- Documentation: ylab-hi.github.io/DeepChopper
- Project page on this site: DeepChopper project
If you find adapter-bridged chimeras in your own data, I’d love to hear about it. The systematic story is only a few cell lines deep right now, and every new dataset adds to the picture of how dRNA-seq actually behaves in the wild.