
1. Introduction#
Noodles and Rust-htslib are two widely used Rust libraries for genomic data handling. While both libraries are designed to work with genomic data, they take different approaches to achieve this goal. This blog explores Noodles and compares it with Rust-htslib, while also discussing its potential pitfalls.
Noodles is a Rust library built on top of Rust’s IO and byte manipulation tools, designed for reading, writing, and manipulating genomic data files. It offers high-level performance and scalability, as well as a high degree of modularity, providing users with many useful tools for working with genomic data.
On the other hand, Rust-htslib is a Rust library that provides a high-level interface to the [HTSlib] C library. It is specifically designed to work with BAM and VCF files, offering a robust set of functions for working with these types of data.
When comparing these two libraries, there are several key differences to consider. Noodles is a more modern library that takes full advantage of Rust’s advanced features, such as iterators and closures. This makes Noodles highly flexible and adaptable to different use cases. Rust-htslib, on the other hand, is a more specialized library designed specifically for working with BAM and VCF files.
2. Usage#
2.1 Use noodles#
The first step is to add noodles as dependencies by using cargo add noodles --featues bam sam bgzf core.
Or we can edit Cargo.toml directly and add the following line:
noodles = {version = "0.32.0", features = ["bam", "sam", "bgzf", "core"]}
2.2 Read bam file#
We can employ the noodles library to read BAM files, and the library offers various methods to access BAM files. What’s more, we can read files asynchronously and process records concurrently. Additionally, it can be combined with the [rayon] library, which offers powerful parallelism features for Rust.
To open a BAM file and read all the records in the file is quite simple:
use noodles::bam;
use noodles::sam;
use std::fs::File;
fn read_bam(path: &str) -> Result<(), Box<dyn std::error::Error>> {
let mut reader = File::open(path).map(bam::Reader::new)?;
let header: sam::Header = reader.read_header()?.parse()?;
reader.read_reference_sequences()?;
reader
.records(&header)
.map(|r| r.unwrap())
.for_each(|record| {
println!("read name: {}", record.read_name().unwrap());
});
Ok(())
}
Before reading records, we need to consume header and reference sequences to direct file handler to the first record. Furthermore, we can read records asynchronously:
use noodles::bam;
use noodles::sam;
use std::fs::File;
fn read_bam_async(path: &str) -> Result<(), Box<dyn std::error::Error>> {
let mut reader = File::open(path).map(bam::Reader::new)?;
let header: sam::Header = reader.read_header()?.parse()?;
reader.read_reference_sequences()?;
reader
.lazy_records()
.map(|r| r.unwrap())
.for_each(|record| {
println!("read name: {}", record.read_name().unwrap().unwrap());
});
Ok(())
}
I utilize hyperfine to conduct benchmarking.
The results show that read_async() is 1.5 times faster than read_bam() when reading the bam file contains 144309 records.
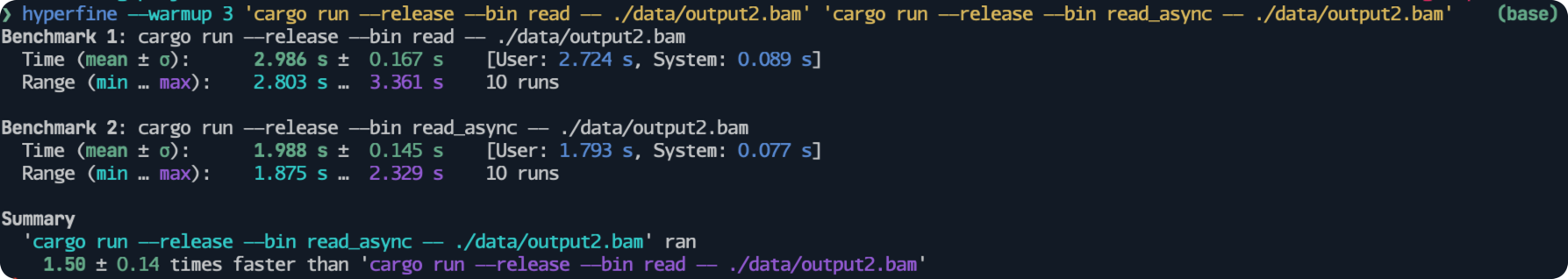
One difference is that we use lazy_records() instead of records(), and read_name().unwrap().unwrap() instead of read_name().unwrap() to get read name.
That is because lazy_records() will return noodles::bam::reader::LazyRecords.
However, records() will return noodles::sam::reader::Records
These two types have different methods, and Records have more methods compared to LazyRecords.
For instance, the cigar object return from LazyRecords is not usable in comparison with cigar object return from Records.
Consequently, we need to reconstruct some data structures from LazyRecords to Records.
For example:
// File: read_bam_async
use anyhow::Context;
use noodles::bam;
use noodles::sam;
use std::fs::File;
use sam::record::cigar::Cigar;
use sam::record::data::Data;
fn read_bam_async(path: &str) -> Result<(), Box<dyn std::error::Error>> {
let mut reader = File::open(path).map(bam::Reader::new)?;
let header: sam::Header = reader.read_header()?.parse()?;
reader.read_reference_sequences()?;
reader
.lazy_records()
.map(|r| r.unwrap())
.for_each(|record| {
let read_name = record.read_name().unwrap().unwrap();
let data = Data::try_from(record.data())
.with_context(|| format!("failed to get data {}", read_name))
.unwrap();
let cigar = Cigar::try_from(record.cigar())
.with_context(|| format!("failed to get cigar {}", read_name))
.unwrap();
let sequence = sam::record::Sequence::try_from(record.sequence())
.with_context(|| format!("failed to get sequence {}", read_name))
.unwrap();
println!("read name: {}, cigar: {}", read_name, cigar);
});
Ok(())
}
The purpose of code is to reconstruct Data, Cigar and Sequence of nsam::record from the data return by LazyRecords.
This is necessary because the data from LazyRecords do not have enough methods to manipulate.
As a result, we can access the tag or field in Data and Cigar through the reconstructed data structure.
Another trick is to convert the Sequence object return by Records or LazyRecords to use noodles::fasta::record::Sequence since we can get reverse complement sequence easily by let rev_comp: Sequence = sequence.complement().rev().collect::<Result<_, _>>()?;
After executing the code block with sample input, we will see following output:

You may notice that we use anyhow::Context here to provide enrich message if there is a bug.
Anyhow is an amazing library, which allow user to handle error more easily.
2.3 Process records in parallel#
Rust is a programming language that enables fearless concurrency.
Its features allow us to safely parallelize programs.
[Rayon] is an excellent Rust library that seamlessly provides parallel iterators.
We can use this library to parallelize our current program and accelerate its execution without too much effort.
Before using Rayon, make sure to add its dependency using cargo add rayon.
// File: read_bam_async_rayon
use anyhow::Context;
use noodles::bam;
use noodles::sam;
use rayon::prelude::*;
use std::fs::File;
use sam::record::cigar::Cigar;
use sam::record::data::Data;
use std::thread::sleep;
fn read_bam_async_rayon(path: &str) -> Result<(), Box<dyn std::error::Error>> {
let mut reader = File::open(path).map(bam::Reader::new)?;
let header: sam::Header = reader.read_header()?.parse()?;
reader.read_reference_sequences()?;
reader
.lazy_records()
.par_bridge() // convert to parallel iterators
.map(|r| r.unwrap())
.for_each(|record| {
let read_name = record.read_name().unwrap().unwrap();
let data = Data::try_from(record.data())
.with_context(|| format!("failed to get data {}", read_name))
.unwrap();
let cigar = Cigar::try_from(record.cigar())
.with_context(|| format!("failed to get cigar {}", read_name))
.unwrap();
let sequence = sam::record::Sequence::try_from(record.sequence())
.with_context(|| format!("failed to get sequence {}", read_name))
.unwrap();
sleep(std::time::Duration::from_millis(1000)); // for benchmarking
println!("read name: {}, cigar: {}", read_name, cigar,);
});
Ok(())
}
For the sake of the benchmarking, I will add another line sleep(std::time::Duration::from_millis(1000)); to simulate labor work.
In this implementation, I am using 4 threads to process an input that has three records.
We obtain a speedup of three times faster than the version without using threads, which is reasonable considering the overhead of launching and joining threads.
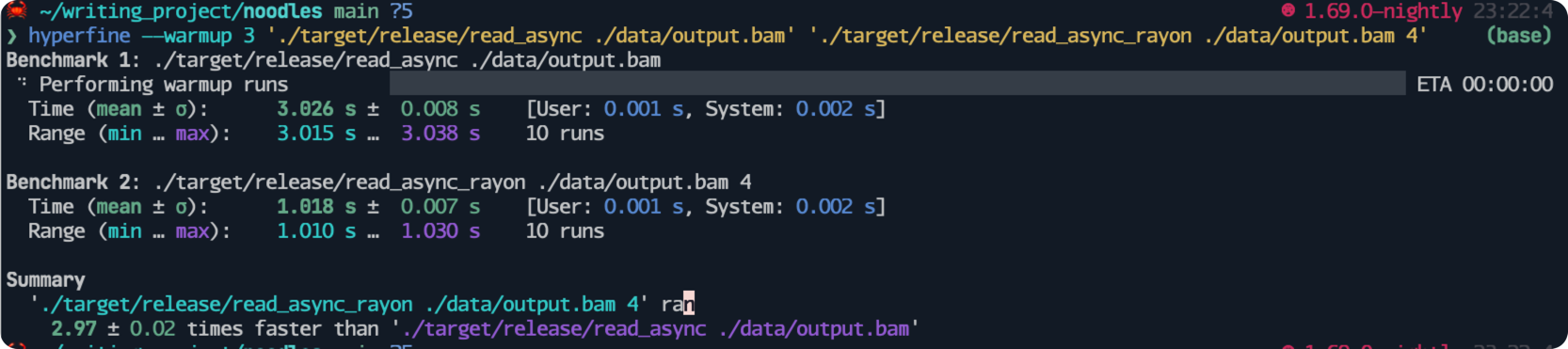
I conducted a benchmarking test for reading records without using asynchronous programming.
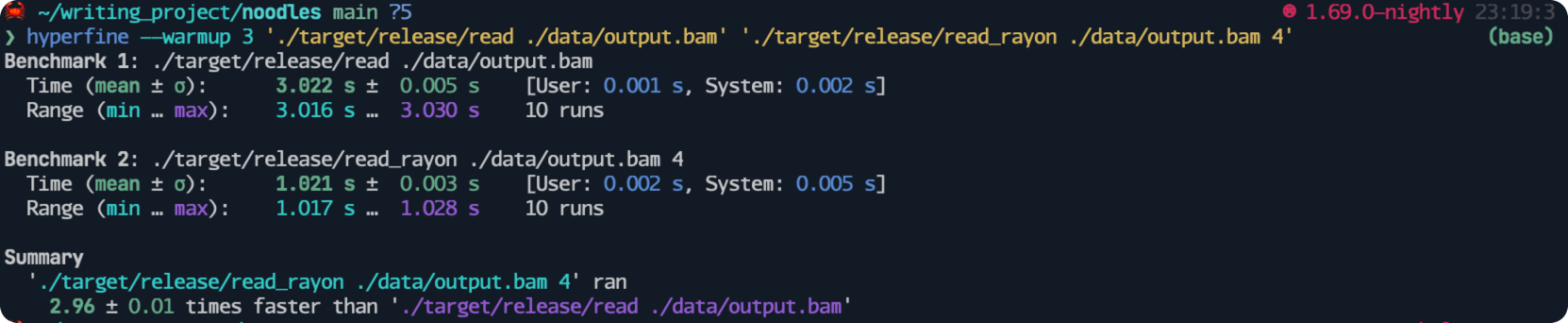
2.4 Query certain region#
An index file is required if you want to query records in specific regions. Similar to rust-htslib, noodles provides a feature to assist users in fetching records from specific regions.
For example,
use anyhow::{Context, Result};
use noodles::bam;
use noodles::sam;
use std::path::Path;
fn query<T>(path: T) -> Result<()>
where
T: AsRef<Path>,
{
let mut reader = bam::indexed_reader::Builder::default()
.build_from_path(path.as_ref())
.with_context(|| {
format!(
"failed to read bam file and index not existed {:?} ",
path.as_ref()
)
})?;
let header: sam::Header = reader
.read_header()
.context("failed to read bam reader")?
.parse()
.context("failed to parse bam rader")?;
let reference = reader
.read_reference_sequences()
.context("failed to read reference sequences")?;
let region = "chr17:79778148-79778149"
.parse()
.expect("failed to parse region");
let count = reader.query(&header, ®ion).unwrap().count();
println!("{} records found", count);
Ok(())
}
fn main() {
let path = std::env::args().nth(1).unwrap();
query(path).unwrap();
}
Please note that the IndexReader assumes that the index file’s name is file_name.bam.bai instead of file_name.bai.
If your index file does not follow this naming convention, you may encounter an error such as file does not exist.
We can also use [rayon] to speed up the code by:
let count = reader.query(&header, ®ion).unwrap().par_bridge().count();
// let count = reader.query(&header, ®ion).unwrap().count();
3. Pitfall#
3.1 Bam/Sam header format#
When utilizing noodles to parse BAM/SAM files, adherence to the standard SAM format is crucial compared to rust-htslib.
Otherwise, parsing may result in errors such as the “Invalid ReadGroup for PL” message.
In this instance, the PL value belonging to the RG tag does not comply with the standard.
According to the standard, PICBIO is one of the correct values to use for PL, with a defined set of values available for it.
A noodles issue has been discussed regarding the strictness of parsing headers.
To resolve this issue, we recommend replacing the RG tag in place using the samtools addreplacerg -r "@RG\tID:test\tSM:hs\tLB:ga\tPL:PACBIO" -w input.bam -o output.bam before processing the file.
Do not forget to index the new file if you want to query certain region.
3.2 IndexReader#
As previously mentioned, using IndexReader to read SAM/BAM files eliminates the need to read the index separately.
However, it is important to note that IndexReader does not expose the same API as Reader, and the data structures for Cigar and Data are different from those used by Reader.
The workaround for this is to reconstruct the relevant data structures used by Reader from those used by IndexReader, as previously mentioned.
3.3 Read file multiple times#
It is important to note that seeking to the first record is necessary when you want to read the file again, but not required when you want to query it again. However, before reading the first record, it is crucial to consume the header and reference to help forward the file handler to the position of the first record. It is not possible to iterate through all the records twice using one file handler since it moves the current file handler to the end of the file.
To overcome this issue, we have two solutions.
Firstly, we can reopen the file handler and consume the header and references respectively.
Secondly, we can move the current file handler to the beginning of the file.
Unfortunately, noodles does not provide an API to do so, and therefore, we need to create our own version based on the unexposed version.
use noodles::bam;
use noodles::sam;
use noodles::bgzf;
use std::io::{self, Read, Seek};
pub trait NoodleBamIndexReaderExt {
fn seek_to_first_record(&mut self) -> io::Result<bgzf::VirtualPosition>;
}
impl<R> NoodleBamIndexReaderExt for bam::indexed_reader::IndexedReader<bgzf::Reader<R>>
where
R: Read + Seek,
{
fn seek_to_first_record(&mut self) -> io::Result<bgzf::VirtualPosition> {
// seek to first record
let areader = self.get_mut();
areader.seek(bgzf::VirtualPosition::default())?;
self.read_header()?;
self.read_reference_sequences()?;
Ok(self.get_ref().virtual_position())
}
}
We have created an extension trait for IndexReader, which enables us to reset the file handler using the seek_to_first_record() method.
For example,
fn count<T>(path: T) -> Result<()>
where
T: AsRef<Path>,
{
let mut reader = bam::indexed_reader::Builder::default()
.build_from_path(path.as_ref())
.with_context(|| {
format!(
"failed to read bam file and index not existed {:?} ",
path.as_ref()
)
})?;
let header: sam::Header = reader
.read_header()
.context("failed to read bam reader")?
.parse()
.context("failed to parse bam rader")?;
let reference = reader
.read_reference_sequences()
.context("failed to read reference sequences")?;
let count1 = reader.lazy_records().count();
println!("first count: {}", count1);
let count2 = reader.lazy_records().count();
println!("second count: {}", count2);
Ok(())
}
The output will be:

After using our extension, it will be changed to:
fn count<T>(path: T) -> Result<()>
where
T: AsRef<Path>,
{
let mut reader = bam::indexed_reader::Builder::default()
.build_from_path(path.as_ref())
.with_context(|| {
format!(
"failed to read bam file and index not existed {:?} ",
path.as_ref()
)
})?;
let header: sam::Header = reader
.read_header()
.context("failed to read bam reader")?
.parse()
.context("failed to parse bam rader")?;
let reference = reader
.read_reference_sequences()
.context("failed to read reference sequences")?;
let count1 = reader.lazy_records().count();
println!("first count: {}", count1);
reader.seek_to_first_record().unwrap(); // important
let count2 = reader.lazy_records().count();
println!("second count: {}", count2);
Ok(())
}
After resetting the file handler, we are now able to iterate over the records as before. However, it is not necessary to do this when fetching records from specific regions. Let’s take a look at the output:

3.4 Off one error#
The noodles library uses a 1-based position and employs a range index syntax that includes both the left and right endpoints, [start, end], to retrieve a sequence.
In contrast, Rust uses a 0-based position and the default range syntax is left-open and right-closed, [start, end).
Therefore, you must add 1 to the starting position when using noodles to retrieve a sequence, otherwise, you may encounter an off-by-one error.
3.5 Get reference name#
In Noodles, it is not intuitive to get reference name.
fn get_reference_name(references: &ReferenceSequences, reference_sequence_id: usize) -> String {
references
.get_index(reference_sequence_id)
.map(|(name, _)| name.as_str())
.unwrap()
.to_owned()
}
We can get reference_sequence_id by record.reference_sequence_id().
4. Conclusion#
One potential issue to consider when using Noodles is its relatively new status in comparison to rust-htslib, which has been available for a longer period of time and is widely used in many projects. As a result, Noodles may contain bugs or other problems that have not yet been discovered. On the other hand, rust-htslib has undergone extensive testing and has proven to be a reliable and high-performance option.
In summary, both Noodles and rust-htslib are valuable Rust libraries for managing genomic data, and each has its own advantages and disadvantages. Choosing between the two depends on the specific needs of the project at hand. Noodles, being a pure Rust implementation, may be the better option if flexibility and adaptability are desired. The sample code can be found at the repository.